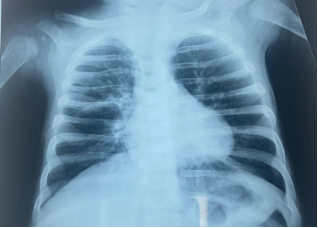
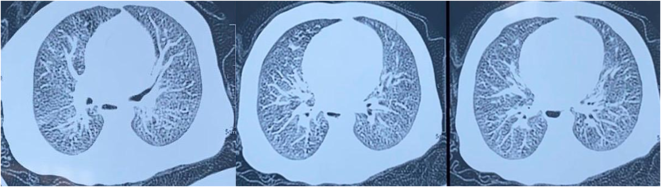
Recurrent wheezing is common in infants, most often triggered by respiratory viral infections. When wheezing begins early and persists without symptom-free intervals, it is crucial to first exclude common underlying conditions such as gastroesophageal reflux disease, bronchopulmonary or vascular malformations, and decompensated cardiac disease. Once these etiologies have been ruled out, less frequent disorders—most notably cystic fibrosis—should be considered. Through this case report, we aim to highlight the diagnostic and management challenges of cystic fibrosis in our setting. A 4-month-old infant was hospitalized for recurrent wheezing and dyspnea without symptom-free intervals. Clinical examination revealed severe respiratory distress with diffuse wheezing, along with growth faltering (–3 SD). Chest X-ray demonstrated perihilar atelectasis, bronchial wall thickening in the left lower lobe and the right apical basal segment, and left-sided thoracic hyperinflation and chest CT angiography showed bilateral pneumonia. Immunological investigations were unremarkable. A sweat chloride test, performed twice, confirmed markedly elevated chloride concentrations. The patient showed a favorable clinical evolution following treatment with antibiotic therapy, pancreatic enzyme replacement, vitamin supplementation, and respiratory physiotherapy. In the absence of systematic neonatal screening in our context and to prevent severe and fatal complications, cystic fibrosis should be considered in any case of persistent wheezing in an infant.
| Published in | American Journal of Pediatrics (Volume 12, Issue 1) |
| DOI | 10.11648/j.ajp.20261201.15 |
| Page(s) | 34-38 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2026. Published by Science Publishing Group |
Cystic Fibrosis, Challenge in the Management, Morocco, Children
CF | Cystic Fibrosis |
CFTR | Cystic Fibrosis Transmembrane Conductance Regulator |
SD | Standard Deviation |
CT | Computed Tomography |
CRP | C-Reactive Protein |
AST | Aspartate Aminotransferase |
ALT | Alanine Aminotransferase |
FEV₁ | Forced Expiratory Volume in One Second |
LU | Lipase Units |
IU | International Units |
DNA | Deoxyribonucleic Acid |
| [1] | Bardin, E.; Pranke, I.; Hinzpeter, A.; Sermet-Gaudelus, I. Traitements de La Mucoviscidose: Révolution Clinique et Nouveaux Défis. Med Sci (Paris) 2024, 40(3), 258-267. |
| [2] | Ravilly, S.; Le Roux, E.; Bellis, G.; Dufour, F. Épidémiologie et Physiopathologie de La Mucoviscidose [Epidemiology and Pathophysiology of Cystic Fibrosis]. Revue Francophone des Laboratoires 2007, 2007(397), 25-36 |
| [3] |
Institut Pasteur. Mucoviscidose [Internet] [Internet]. Disponible sur:
https://www.pasteur.fr/fr/centre-medical-fiches/fiches-maladies/mucoviscidose |
| [4] |
Inserm. Mucoviscidose: Des pistes thérapeutiques encourageantes. Inserm - La science pour la santé. Available from:
https://www.inserm.fr/information-en-sante/dossiers-information/mucoviscidose (accessed 15 January 2026). |
| [5] |
Tomaiuolo R, Spina M, Castaldo G. Molecular Diagnosis of Cystic Fibrosis: Comparison of Four Analytical Procedures. Clinical Chemistry and Laboratory Medicine [Internet]. 27 janv 2003 [cité 27 févr 2025]; 41(1). Disponible sur:
https://www.degruyter.com/document/doi/10.1515/CCLM.2003.006/html |
| [6] | Jaouad IC, Elalaoui SC, Sbiti A, Elkerh F, Belmahi L, Sefiani A. CONSANGUINEOUS MARRIAGES IN MOROCCO AND THE CONSEQUENCE FOR THE INCIDENCE OF AUTOSOMAL RECESSIVE DISORDERS. J Biosoc Sci. sept 2009; 41(5): 575‑81. |
| [7] | Ratbi I, Génin E, Legendre M, Le Floch A, Costa C, Cherkaoui-Deqqaqi S, et al. Cystic fibrosis carrier frequency and estimated prevalence of the disease in Morocco. Journal of Cystic Fibrosis. sept 2008; 7(5): 440‑3. |
| [8] | Hubert, D.; Le Bourgeois, M. Atteinte respiratoire de la mucoviscidose de l’enfance à l’âge adulte. Archives de Pédiatrie 2012, 19, S17-S19. |
| [9] | Emerson J, Rosenfeld M, McNamara S, Ramsey B, Gibson RL. Pseudomonas aeruginosa and other predictors of mortality and morbidity in young children with cystic fibrosis. Pediatric Pulmonology. août 2002; 34(2): 91‑100. |
| [10] | Sarles J. Atteinte digestive (pancréatique et intestinale) de la mucoviscidose : approche physiopathologique. Archives de Pédiatrie. mai 2012; 19: S20‑2. |
| [11] | Kessler, L.; Abély, M. Atteinte pancréatique exocrine et endocrine dans la mucoviscidose. Archives de Pédiatrie 2016, 23(12), 12S21-12S32. |
| [12] | Farrell PM, Kosorok MR, Rock MJ, Laxova A, Zeng L, Lai HC, et al. Early Diagnosis of Cystic Fibrosis Through Neonatal Screening Prevents Severe Malnutrition and Improves Long-Term Growth. Pediatrics. 1 janv 2001; 107(1): 1‑13. |
| [13] | Mishra A, Greaves R, Massie J. The relevance of sweat testing for the diagnosis of cystic fibrosis in the genomic era. Clin Biochem Rev. nov 2005; 26(4): 135‑53. |
| [14] | Dasenbrook EC, Konstan MW, VanDevanter DR. Association between the introduction of a new cystic fibrosis inhaled antibiotic class and change in prevalence of patients receiving multiple inhaled antibiotic classes. Journal of Cystic Fibrosis. mai 2015; 14(3): 370‑5. |
| [15] | Lahzami S, Nicod LP. [Inhaled therapies for cystic fibrosis]. Rev Med Suisse. 23 nov 2011; 7(318): 2285‑8. |
| [16] | McIlwaine M. Chest physical therapy, breathing techniques and exercise in children with CF. Paediatric Respiratory Reviews. mars 2007; 8(1): 8‑16. |
| [17] | Struyvenberg MR, Martin CR, Freedman SD. Practical guide to exocrine pancreatic insufficiency - Breaking the myths. BMC Med. 10 févr 2017; 15(1): 29. |
| [18] | Abdallah K, De Boeck K, Dooms M, Simoens S. A Comparative Analysis of Pricing and Reimbursement of Cystic Fibrosis Transmembrane Conductance Regulator Modulators in Europe. Front Pharmacol. 8 nov 2021; 12: 746710. |
| [19] | Fajac I, Burgel PR, Martin C. New drugs, new challenges in cystic fibrosis care. Eur Respir Rev. juill 2024; 33(173): 240045. |
APA Style
Merroun, I., Alaoui-Inboui, F. Z., Ahmito, O., Achiwi, M., Chbani, K., et al. (2026). Challenges in the Management of Cystic Fibrosis in Morocco: A Case Report. American Journal of Pediatrics, 12(1), 34-38. https://doi.org/10.11648/j.ajp.20261201.15
ACS Style
Merroun, I.; Alaoui-Inboui, F. Z.; Ahmito, O.; Achiwi, M.; Chbani, K., et al. Challenges in the Management of Cystic Fibrosis in Morocco: A Case Report. Am. J. Pediatr. 2026, 12(1), 34-38. doi: 10.11648/j.ajp.20261201.15
@article{10.11648/j.ajp.20261201.15,
author = {Ibtissam Merroun and Fatima Zahra Alaoui-Inboui and Othmane Ahmito and Meriem Achiwi and Kamelia Chbani and Bouchra Slaoui},
title = {Challenges in the Management of Cystic Fibrosis in Morocco: A Case Report},
journal = {American Journal of Pediatrics},
volume = {12},
number = {1},
pages = {34-38},
doi = {10.11648/j.ajp.20261201.15},
url = {https://doi.org/10.11648/j.ajp.20261201.15},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ajp.20261201.15},
abstract = {Recurrent wheezing is common in infants, most often triggered by respiratory viral infections. When wheezing begins early and persists without symptom-free intervals, it is crucial to first exclude common underlying conditions such as gastroesophageal reflux disease, bronchopulmonary or vascular malformations, and decompensated cardiac disease. Once these etiologies have been ruled out, less frequent disorders—most notably cystic fibrosis—should be considered. Through this case report, we aim to highlight the diagnostic and management challenges of cystic fibrosis in our setting. A 4-month-old infant was hospitalized for recurrent wheezing and dyspnea without symptom-free intervals. Clinical examination revealed severe respiratory distress with diffuse wheezing, along with growth faltering (–3 SD). Chest X-ray demonstrated perihilar atelectasis, bronchial wall thickening in the left lower lobe and the right apical basal segment, and left-sided thoracic hyperinflation and chest CT angiography showed bilateral pneumonia. Immunological investigations were unremarkable. A sweat chloride test, performed twice, confirmed markedly elevated chloride concentrations. The patient showed a favorable clinical evolution following treatment with antibiotic therapy, pancreatic enzyme replacement, vitamin supplementation, and respiratory physiotherapy. In the absence of systematic neonatal screening in our context and to prevent severe and fatal complications, cystic fibrosis should be considered in any case of persistent wheezing in an infant.},
year = {2026}
}
TY - JOUR T1 - Challenges in the Management of Cystic Fibrosis in Morocco: A Case Report AU - Ibtissam Merroun AU - Fatima Zahra Alaoui-Inboui AU - Othmane Ahmito AU - Meriem Achiwi AU - Kamelia Chbani AU - Bouchra Slaoui Y1 - 2026/03/17 PY - 2026 N1 - https://doi.org/10.11648/j.ajp.20261201.15 DO - 10.11648/j.ajp.20261201.15 T2 - American Journal of Pediatrics JF - American Journal of Pediatrics JO - American Journal of Pediatrics SP - 34 EP - 38 PB - Science Publishing Group SN - 2472-0909 UR - https://doi.org/10.11648/j.ajp.20261201.15 AB - Recurrent wheezing is common in infants, most often triggered by respiratory viral infections. When wheezing begins early and persists without symptom-free intervals, it is crucial to first exclude common underlying conditions such as gastroesophageal reflux disease, bronchopulmonary or vascular malformations, and decompensated cardiac disease. Once these etiologies have been ruled out, less frequent disorders—most notably cystic fibrosis—should be considered. Through this case report, we aim to highlight the diagnostic and management challenges of cystic fibrosis in our setting. A 4-month-old infant was hospitalized for recurrent wheezing and dyspnea without symptom-free intervals. Clinical examination revealed severe respiratory distress with diffuse wheezing, along with growth faltering (–3 SD). Chest X-ray demonstrated perihilar atelectasis, bronchial wall thickening in the left lower lobe and the right apical basal segment, and left-sided thoracic hyperinflation and chest CT angiography showed bilateral pneumonia. Immunological investigations were unremarkable. A sweat chloride test, performed twice, confirmed markedly elevated chloride concentrations. The patient showed a favorable clinical evolution following treatment with antibiotic therapy, pancreatic enzyme replacement, vitamin supplementation, and respiratory physiotherapy. In the absence of systematic neonatal screening in our context and to prevent severe and fatal complications, cystic fibrosis should be considered in any case of persistent wheezing in an infant. VL - 12 IS - 1 ER -
Pneumology and Allergology Paediatric Department, Abderrahim Harrouchi Mother and Child Hospital, Casablanca, Morocco
Pneumology and Allergology Paediatric Department, Abderrahim Harrouchi Mother and Child Hospital, Casablanca, Morocco